Background
Background
Cervical pain from spondylosis or muscular problems is a very common symptom in the general adult population, estimated in a recent study to have a point prevalence of 4.9% and a global burden of 33.6 million disability-adjusted life years (Hoy et al., 2014). Symptoms are commonly recurrent within individuals, returning in from 50-85% of cases within 5 years of initial presentation (Haldeman et al., 2008).
The most common aetiologies of cervical pain are joint disease resulting in spondylosis and acute or chronic muscular injury. The muscles of the mobile cervical and lumbar spine tend to develop spasm as a consequence of joint or muscle inflammation and this further exacerbates injury, resulting in a vicious cycle of pain.
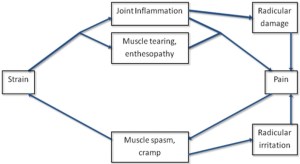
Vicious cycle of neck pain
Diagnosis and management of cervical pain is often complicated by a number of associated symptoms, including headache, dizziness, tinnitus and ear discomfort (Baron et al., 2011). While headache of tension type character, often occipital and radiating anteriorly to the frontalis or temporalis areas, is well-established in relation to neck pain with around 20% of cases of chronic headache having a cervical basis, the other associated symptoms are more controversial.
This article focuses on one associated symptom in particular, namely cervical vertigo. The review article by Brandt and Bronstein, published in JNNP in 2001 presents a comprehensive account of the scientific basis underlying the condition, and in the article I will go over this review and more recent studies on the subject.
Cervical Vertigo Definition and Terminology
Cervical vertigo may be defined as:
A perception of vertigo or imbalance resulting from cervical spondylosis and muscle spasm, normally a chronic tendency to brief attacks and especially brought on by head movement.
Other terms that describe the same thing are cervicogenic vertigo, cervical imbalance and cervical dizziness.
Vertigo arising from the neck presents a particular diagnostic challenge as other potential causes may require alternative management and in some cases will require urgent attention (table 1); the common occurrence of cervical pain means its association with imbalance may be coincidental.
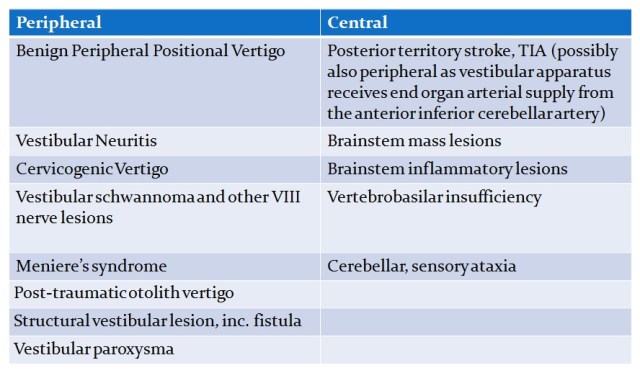
Table 1. Differential diagnosis of vertigo
Diagnostic Confusion
The two conditions most commonly confused with cervical vertigo are benign peripheral positional vertigo (BPPV) and vertebrobasilar insufficiency. They all typically present with vertigo whenever the head moves in a certain way suddenly, rather than a discrete episode as in Meniere’s syndrome, and there are no other cranial nerve features as may occur with a vestibular Schwannoma. Nowadays the availability of MR imaging means that the latter may have already been excluded before specialist referral. The distribution of cases of such vertigo between these three apparently nosologically and pathophysiologically distinct entities remains controversial, and indeed some argue that cervical vertigo does not exist at all.
In truth, the conditions themselves may overlap. Patients with cervicogenic vertigo are likely to have cervical spondylosis making them susceptible also to vertebrobasiliar insufficiency, and if the latter is demonstrated clinicians may be tempted to consider it the hierarchically dominant or sole diagnosis even though many of a patient’s attacks may be cervicogenic. Conversely, patients with BPPV are likely to stiffen their neck as a protective mechanism and may consequently develop cervicogenic vertigo even after their BPPV has resolved. Finally, in post-traumatic cases vertigo may result from a combination of dislodgement of otoconia into the lumen of the semi-circular canals producing BPPV, damage to the otolith organs which are vulnerable to mechanical acceleration, and whiplash injury producing cervicogenic vertigo.
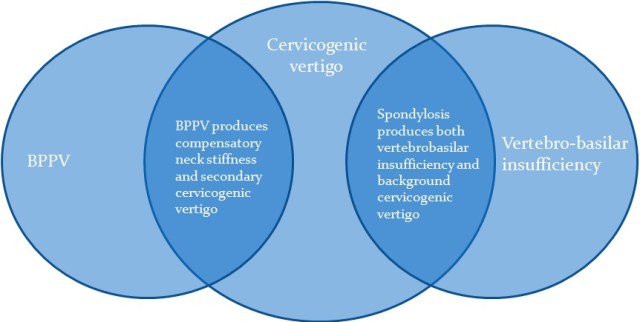
Overlapping nosological entities of vertigo
Clinical Presentation of Cervical Vertigo
When considering the complaint of “dizziness”, it is important to define more closely what the patient actually experiences. When many patients describe dizziness they are actually referring to presyncopal symptoms. True vertigo is a perception that the environment is moving in a rotatory direction or swaying to and fro. Finally, dizziness is sometimes used to describe a spatial disorientation or perception of imbalance. Sometimes it is the perception that appears in itself to be pathological – the patient feels unsteady perhaps more than actually being unsteady; this must be distinguished from patients whose perception of imbalance is a relatively more accurate and objective appraisal of their actual unsteadiness as a result of ataxia or loss of postural reflexes.
The typical patient with cervical vertigo falls into the category of those who describe a perception of spatial disorientation or imbalance. Rather than true vertigo, they report positional unsteadiness, imbalance, giddiness, or a feeling that the ground is sliding underneath them. As is typical for peripheral vertigo, head movements precipitate the symptoms, often neck extension or rising from supine. They may have an excessively cautious gait for their apparent objective level of balance impairment, grabbing hold of walls or “furniture walking”, and this may lead to a mistaken diagnosis of psychogenic vertigo. For such symptoms to be considered cervicogenic, there should obviously be a history of neck pain. However many specialists recommend caution in making a diagnosis of cervicogenic vertigo whenever there is neck pain, especially in cases where the description is of true vertigo. The musculature is obviously not the only structure in the neck that can give neck pain and vertigo and if post traumatic vertebral artery dissection must be excluded. Similarly, vertebrobasilar insufficiency (see below) may result from pinching of the arteries in their course through the cervical vertebrae and, while this would typically also result in other transient brainstem symptoms, a presentation with vertigo alone has been reported.
On examination, patients with cervical vertigo sometimes have their symptoms set off on testing eye movements, and they may have a reluctance or a restriction on testing range of head movement. Instead they may tend to turn the trunk with the neck. Vestibulo-ocular reflex testing or the Hallpike’s test may reproduce symptoms but without nystagmus or with a very slight nystagmus. Instead, to isolate the influence of cervical afferents the patient should be placed on a rotating stool and the head gently fixed by the examiner’s hands while the trunk is rotated back and forth. Cervical nystagmus of immediate onset may result, changing direction with the direction of rotation. However, this sign is unreliable, as discussed below.
Important Alternative Diagnosis: Benign Peripheral Positional Vertigo
Bingin peripheral positional vertigo (BPPV) is thought to relate to otoconia floating freely in the semicircular canals, usually the posterior semicircular canal on one side; head movement in the plane of this canal results in ongoing stimulation and generates vertigo and nystagmus.
Onset of BPPV is usually subacute or chronic, and characterised by brief episodes on making certain head movements. A more severe acute onset of continuous positional vertigo usually points instead to vestibular neuritis, also called labyrinthitis.
The Hallpike’s test is positive in BPPV, with rotatory nystagmus in an extorting direction in the lower eye (top of the eyes jerking towards the floor) and usually adapting after several seconds.
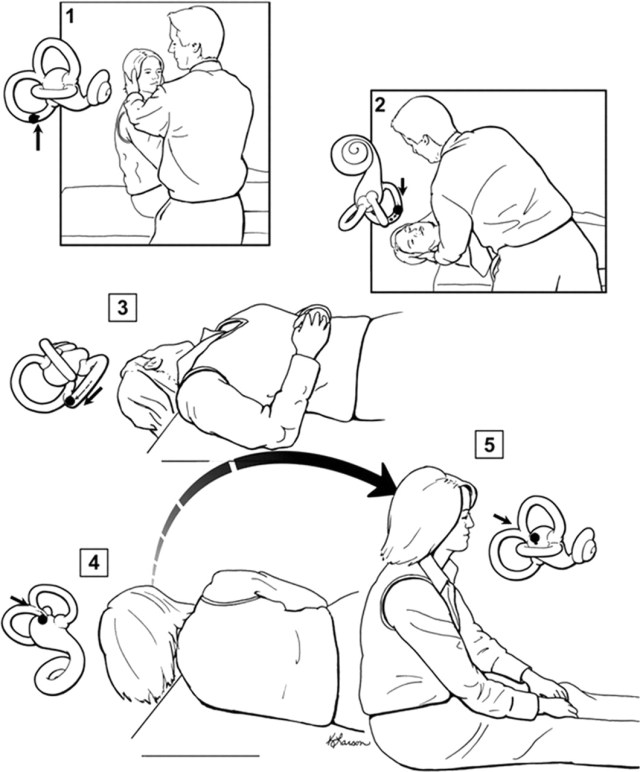
Hallpike’s Test and Epley Manoeuvre (Fife et al., 2008). Otoconia are loose in the right posterior semicircular canal (arrowed fig. 1). The patient’s head is turned 45 degrees to the right so that the posterior canal is in the plane of motion (and the effect of the posterior canal on the other side is negated) when the patient is lain flat (fig. 2). The consequent nystagmus is extorting in the right eye.
Vertebrobasilar Insufficiency
This is an oft cited but rarely demonstrated syndrome thought to relate to pinching of the ipsilateral vertebral artery when a patient with cervical spondylosis turns their neck. The tortuous course of the artery through the transverse foramina of cervical vertebrae C6 to C1 and then across the posterior arch of C1 makes the artery particularly susceptible to such compression.
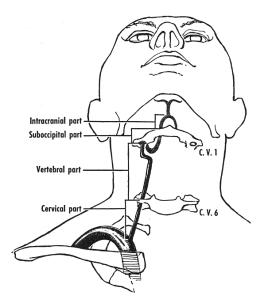
Course of the vertebral artery
In the related Barré Liéou syndrome (1926), the artery is not directly pinched but irritation of the sympathetic plexus around vertebral arteries causes reflex vasoconstriction. However, the existence of this sympathetically mediated phenomenon remains doubtful.
On examination, it is suggested that if the head is moved to one extreme, compared to cervical vertigo, the nystagmus of vertebrobasilar insufficiency would start only after a delay of several seconds to minutes. However, given it is assumed that a perhaps already atherosclerotic artery is being badly pinched, I cannot help but think that such a test should only be performed in a catheter lab! Indeed, it would only be during formal arterial angiography demonstrating occlusion with the head turned to one side but not the other coul done really make a confident diagnosis of this condition.
It is considered by some that the phenomeon of disruption of cervical afferents mediating the cervico-ocular reflex does not exist and “cervicogenic vertigo” always results from vertrbrobasilar insufficiency. However, a review of the anatomy of the blood supply to the brain via the circle of Willis reveals many collaterals and means that ischaemia will result only when there is already occlusion of the contralateral artery, and probably additional significant atheromatous narrowing of the anterior circulation.
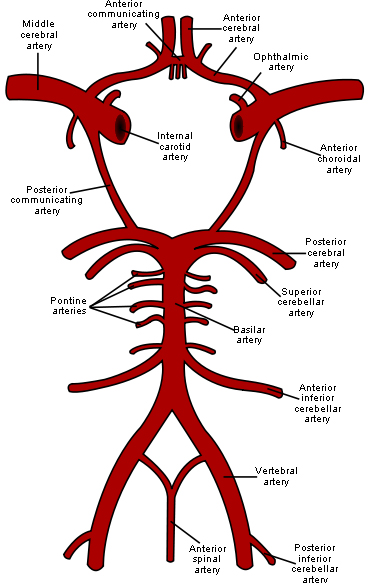
Circle of Willis
Such a situation must be rare and is different of course from the aetiology of a vertebrobasilar territory transient ischaemic attack, where an embolus from these arteries passes up and lodges into a smaller artery without collaterals. This is why vertebral or carotid artery occlusion in the neck carries a much lower risk of stroke than does a stenosis where emboli may still pass up through the narrowed lumen.
In addition, if there was transient ischaemia from hypoperfusion, why would it selectively result in vertigo and no other brainstem features such as ataxia, dysarthria, collapse, hemianopia or loss of consciousness? Nevertheless, some cases of likely vertebrobasilar insufficiency have been reported to present with vertigo without other brainstem features (Dvorak & Dvorak, 1990). Conceivably if the blood supply to an already stenosed anterior inferior cerebellar artery, which comes off the basilar artery not the vertebral artery, was critically dependent on one remaining patent vertebral artery, pinching of the latter could result in transient ischamia only of the inner ear structures, resulting in a peripheral nystagmus with or without deafness and tinnitus.
The main point is that while vertebrobasilar insufficiencty may indeed exist, the circumstances required for it to occur seem too rare to account for all cases of presumed cervical vertigo.
Scientific Basis of Cervicogenic Vertigo
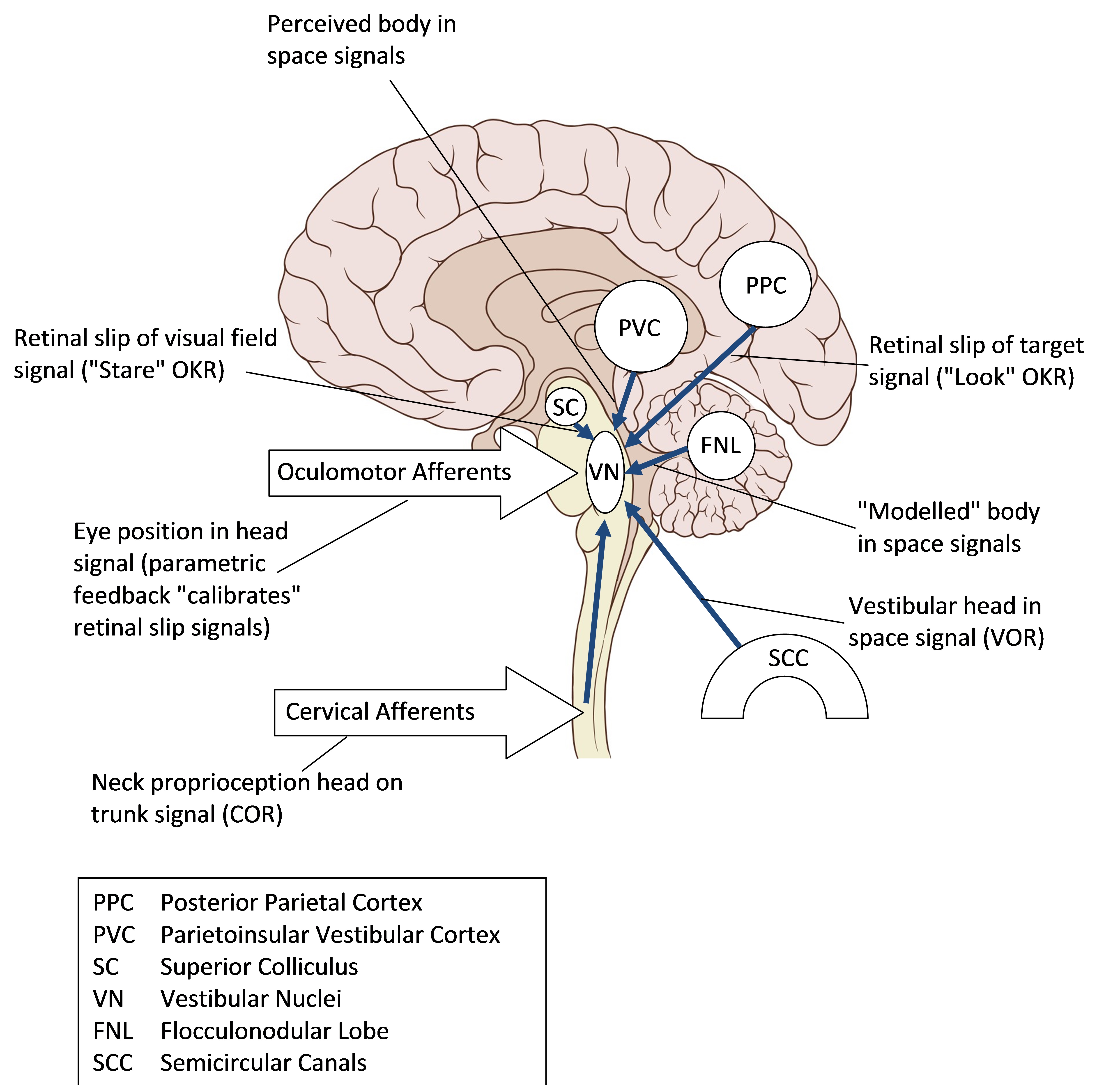
Signals important in balance control, including vision, eye position, vestibular signals and processed postural information and perceptual information, are integrated in the vestibular nuclei located in the pons. These nuclei in turn output to postural control centres, to the eye movement apparatus to control compensatory eye movements and to perceptual processes.
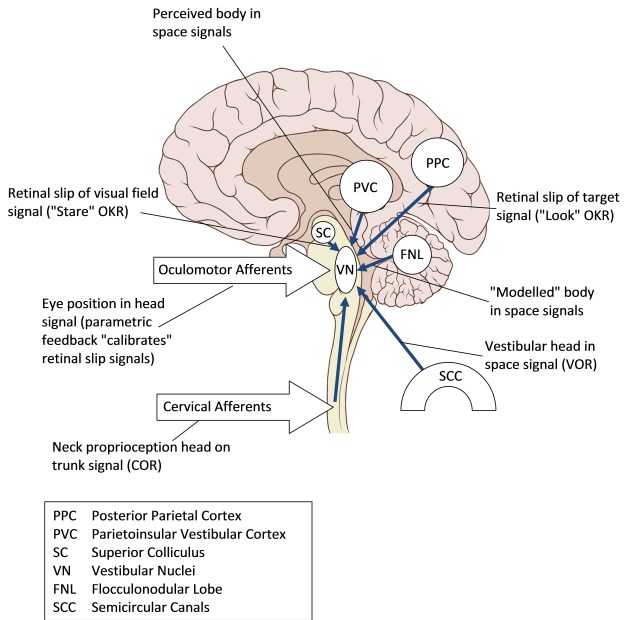
Inputs to the Vestibular Nuclei
Vertigo is a false perception of movement, and typically results not from a deficit but a mismatch of balance signals. This mismatch may be between defective and normal vestibular canal signals on either side of the head, or between vestibular and visual signals. For cervical vertigo to exist, there would therefore have to be a physiological basis not only in functionally important cervical signals inputting head position on the trunk to the vestibular nuclei but also in a process that compared these signals with vestibular or visual signals at a perceptual level so that a mismatch could lead to vertigo.
Why have cervical signalling for balance?
Visual inputs signal movement and position relative to the retina, while vestibular inputs signal movement and position relative to the head; however, the balance system needs information primarily on the centre of mass which mainly reflects the trunk. In essence, afferents from muscle spindles and joint receptors in the neck would allow determination of centre of mass by correction of the vestibular signal for head position with respect to the trunk.
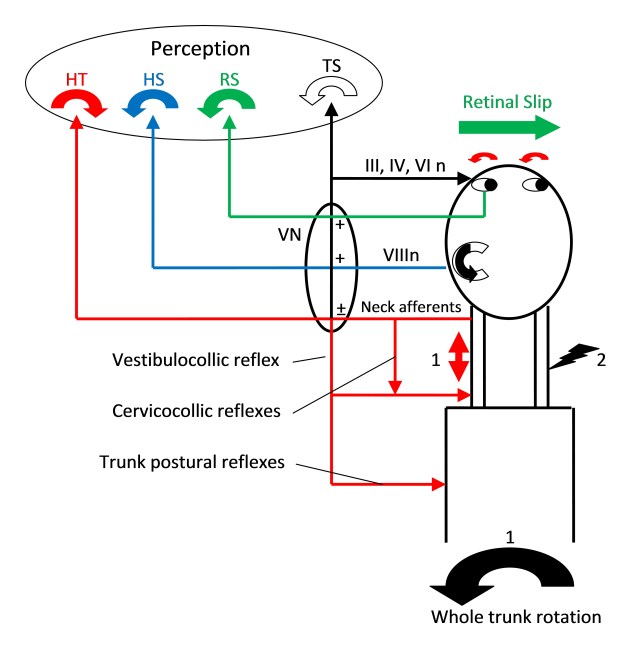
Balance Responses.
1) Whole trunk movement to left may leave head behind from inertia. Stretch of left neck muscles signals head on trunk movement to right (red). Lateral semicircular canal signals partial head movement to left (blue), as does retinal slip signal (green). The cervico-ocular reflex (COR) will result in slow phase eye movement to left. This acts to compensate for relative head movement to fix gaze, or shift gaze to overall body facing. Any actual left head movement will result in vestibule-ocular reflex (VOR) slow phase to right and optokinetic reflex (OKR) slow phase to right. Overall eye movement will be integrated in the vestibular nuclei as the difference between COR and an amalgam of VOR and OKR. The separate head on trunk, head in space and retina in space movements may reach level of perception. For an overall perception of trunk motion, leftward head perception, must be added to rightward head on neck perception (which reflects a leftward trunk under head movement). Other reflexes in action include direct cervico-collic stretch reflexes that will turn the head left in response to head on trunk movement, and from the vestibular nuclei an integrated vestibulo-collic reflex that will stabilise the head on the trunk and integrated postural reflexes that will stabilise trunk positioning.
2) Experimental blocking of afferents of right neck will lead to unopposed stretch signalling on left, simulating right head on trunk motion. This will generate an unopposed COR signal slow phase eye movement to left, so fast phase of spontaneous nystagmus is on the same side as the block.
3) Vibration applied to the neck muscle stimulates stretch reflexes without any vestibular or ocular involvement (unless the stretch actually secondarily moves the head).
Cervico-Ocular Reflex Nystagmus
In the same way as vestibular signals are responsible for vestibulo-ocular reflexes and vestibular vertigo, a functionally important cervical balance pathway that could result in cervicogenic vertigo might be expected to be associated with a demonstrable cervico-ocular reflex, where stimulation of spindle afferents results in reflexive compensatory eye movements. In other words, neck proprioception, if input to vestibular nuclei, may result not only in perception of motion, but a compensatory eye movement that may be recorded by electronystagmography, infrared or video systems.
Trunk rotation, e.g. to left, under a fixed head in the dark would be interpreted by neck proprioceptors as head movement to right and would generate a compensatory slow phase to left. Fast phase of nystagmus would therefore be to the right, the opposite side to trunk rotation.
More physiologically, if the trunk turned and the head was not fixed but lagged behind by inertia, the reflex would make the direction of gaze follow the direction of trunk movement even if the head did not.
Evidence for Cervical Balance Signals: Anatomical Connections
The deep short intervertebral neck muscles are rich in muscle spindle afferents that are able to provide a signal of head on trunk position or head on trunk movement (Cooper & Daniel, 1963) and there is anatomical demonstration of connectivity to the vestibular nuclei and neighbouring brainstem reticular formation areas (Ciriani et al., 1992).
Evidence for Cervical Balance Signals: Vibration Induced Responses
Selective stimulation of cervical afferents by vibration over the neck muscles simulates a stretch reflex; unilateral stimulation is indeed found to result in postural responses, apparent movement of a visual target and a weak deviation of perception of subjective vertical to give the illusion of ipsilateral head tilt.
There is also an associated cervico-ocular reflex of low amplitude. Furthermore it is found that this response is increased after a unilateral vestibular lesion, building up over several weeks as a presumed compensatory enhancement. The automatic postural responses are greater than the perception of motion, unlike the major perception of motion that results from caloric testing of vestibular function, and thus fits with cervicogenic vertigo constituting more a sense of imbalance than actual vertigo.
Evidence for Cervical Balance Signals: Disruption of Cervico-Ocular Reflexes
Interference with cervical afferents in an attempt to mimic the situation in cervicogenic vertigo also yields unclear results. Local anaesthesia of the deep neck muscles in humans results in gait deviation, a tendency to fall with a positive Romberg test to the injected side, a perception of altered position and an unsteadiness on sudden head movement that lasts for several hours after the injection. These findings are confirmed on therapeutic C2 level anaesthetic block to treat patients with cervicogenic headache.
However there is no associated nystagmus (ie cervico-ocular reflex), nor any actual vertigo. Some of the effects could reflect an imbalance in muscle tone as a result of cervicocollic reflexes rather than the cervico-ocular reflex. Nevertheless, the pattern fits with the perception of imbalance or “quasi-vertigo” on head movement rather than the true vertigo of vestibular dysfunction.
Evidence for Cervical Balance Signals: Physiological Responses
As described above clinically, on testing trunk rotation under fixed head in the dark, there is sometimes a weak cervico-ocular reflex. However, if the head is not fixed, there may be head movement, limited by inertia but brought on by tissue elasticity and by cervico-collic reflexes. Any actual movement will result in vestibular ocular reflexes and vestibulo-collic reflexes that will secondarily stabilise the head. If tthe head strongly fixed, perceptual processes or pressure detection on the side of the head may also suppress any illusion of head rotation.
It is therefore not surprising that physiological stimulation of putative cervico-ocular reflexes in normal human adults using trunk movements with a stabilised head produces less clear perception of motion than does muscle vibration and unreliable cervico-ocular reflexes. Nevertheless, under carefully controlled conditions, such as sinusoidal trunk movements with the head fixed by a bite bar, a reliable cervico-ocular response can be recorded and compared with analoguos vestibular and optokinetic responses.
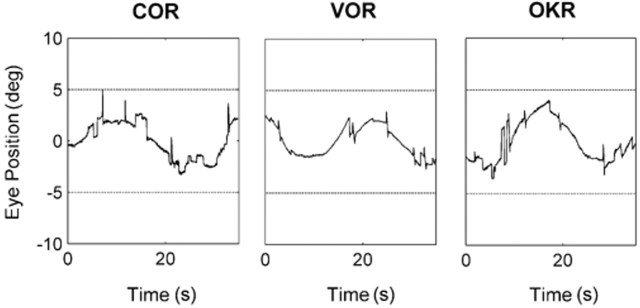
Infra-red recordings of cervico-ocular reflex, vestibulo-ocular reflex and optokinetic reflex resulting from sinusoidal movements (0.04 Hz, ± 5° amp). This isolates the slow phase component as there is no need for resetting saccades of nystagmus when tracking a back and forth sinusoid. (Kelders et al., 2003)
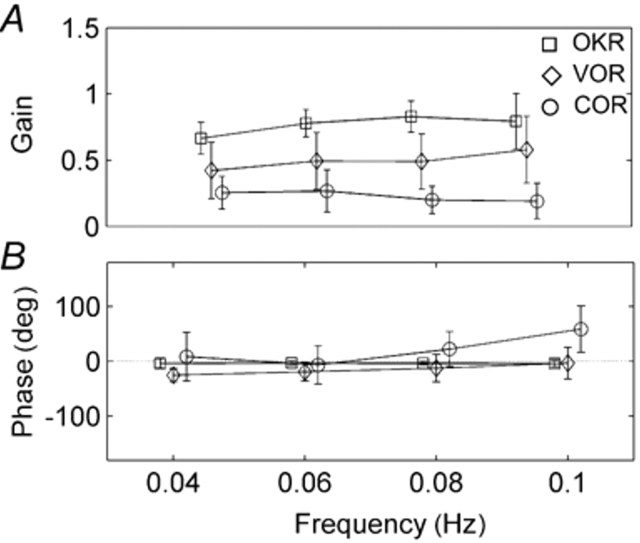
Mean amplitudes of reflex responses at different sinusoid stimulus frequencies. Gain of COR is lowest (VOR low at slow frequencies but increases with higher frequencies).
Phase of VOR and COR are more variable and COR lags behind trunk rotation at higher frequencies. With old age, VOR and OKN gain decrease; there is a compensatory increase in COR gain, as there is after vestibular dysfunction.
Sinusoidal movements of slow frequency and small amplitude generate cervico-ocular reflex (COR) of lower gain than VOR and OKN, and a tendency to lag behind the movement at higher frequencies. With old age, VOR and OKN gain decrease but there is a compensatory increase in COR gain, as there is after vestibular dysfunction. It is tempting to speculate that the same might apply to patients with clincal cervical vertigo.
Studies on patients with cervical vertigo
Having experimentally demonstrated the functioning of cervical balance signals in normal subjects, the next step is to demonstrate disordered signalling in patients with presumed cervical vertigo.
Such patients do have myofascial trigger points for their pain that exhibit spontaneous EMG activity compatible with hyperactive muscle spindles (Hubbard & Berkoff, 1993). However, no correlation is found between the magnitude of physiological cervico-ocular reflexes and the severity of clinical cervicogenic vertigo. Perhaps, if there is an abnormality of cervico-ocular reflexes associated with cervical vertigo, it does not simply relate to the gain (amplitude) of what is after all a physiological rather than pathological phenomenon, but to a mismatching of signalling from either side or to a failure to calibrate such signals with vestibular and visual balance information.
What has been reported in patients with cervical pain (with or without vertigo) is that they tend to have poorer postural control based on vibration or galvanically induced body sway and when such patients are treated with physiotherapy there is improvement in their dizziness and imbalance as well as in their cervical pain (Karlberg et al., 1996).
Does Cervical Vertigo Exist?
There appears to be a scientific basis for the notion that stretch of neck muscles influences balance mechanisms, and a physiological cervico-ocular reflex, especially in controlled conditions, has been demonstrated. However, there has as yet been no demonstrated abnormality in this reflex in patients with cervical vertigo. This lack of a reliable diagnostic test, unlike the clear abnormality of vestibulo-ocular reflexes in vestibular vertigo, hampers study of the condition because of the consequent problem with defining a patient population.
Given the lack of any diagnostic abnormality in such patients other their associated neck pain, the question may then be asked why all patients with neck pain do not get vertigo. This has been felt to signify that the condition does not actually exist. However, there are innumerable examples in medicine where patients do not have to have the “full house” of clinical features to have a syndrome. There may be additional factors that trigger vertigo, such as the nature and asymmetry of muscle spasm, previous vestibular problems or a constitutional tendency to heightened vertiginous perceptions as is found in visual vertigo. Rather than proof the condition does not exist, it might be more constructive to consider patients with neck pain without vertigo a good control population. Using healthy subjects as controls runs the risk of identifying abnormalities that are more the direct result of pain than a manifestation of cervical vertigo.
Since physiotherapy appears to help cervical vertigo as well as pain, it might be regarded that the diagnosis is somewhat pointless since management is the same as if a patient presented with pain alone. However, there is an important differential diagnosis of vertigo that could be occurring coincidental to cervical spondylosis. And management may be the same precisely because the condition is poorly defined. There could be future refinements of physiotherapy techniques if the subset of patients with neck pain who also have vertigo were better understood.
As mentioned above, the contrast with vestibular vertigo where there is an obvious abnormality of vestibule-ocular reflexes, is one factor that has thrown doubt upon the entitiy of cervical vertigo. However, this contrast should in fact be expected given that patients with cervical vertigo actually complain of imbalance and vague giddiness more than true vertigo. Part of the problem with recognising cervical vertigo as a nosological entity may be that the term itself is a misnomer: cervical imbalance may be a more accurate name, serving to remind clinicians that spasm of neck muscles may result in imbalance through cervico-collic as well as cervico-vestibular pathways and that true vertigo in the context of neck pain bears further investigation of other causes of vertigo where the spondylosis is coincidental.
Other Symptoms Associated with Cervical Spondylosis
I have commonly found in clinical practice that certain symptoms often cluster together in the same individuals. In the presence of cervical spondylosis, there seems more than a chance occurrence not only of vertigo, but headache and tinnitus. While it could be argued that their vertigo is always benign peripheral positional vertigo, their headache is migraine and their tinnitus is from cochlear degeneration or “functional”, Occam’s razor and common sense encourages us to look for a single unifying cause.
I tend to call these associations with cervical spondylosis CHIT syndrome (Cervical Headache, Imbalance and Tinnitus).
Headache is well-described in association with cervical spondylosis, and this review has discussed the likely association with vertigo. Tinnitus seems a very unlikely association, but has in fact previously been cited as being linked with cervical vertigo (Brown, 1992) or abnormalities in the neck muscles (Reisshauer et al., 2006).
It initially seems bizarre how an auditory symptom could be associated with spondylosis. However, there is an interesting physiological phenomenon of muscle contraction called the Piper rhythm. When muscles contract steadily and strongly, their individual motor units tend to fire synchronously in a tuned rhythm at around 40 Hz so that the whole muscle vibrates at this frequency. This frequency, and most likely harmonics thereof, can actually be heard by placing a stethoscope over the belly of the contracting muscle. It can be demonstrated using frequency analysis of electromyogram, tremor and electronic stethoscope signals that what is heard does indeed relate to this motor unit activity.
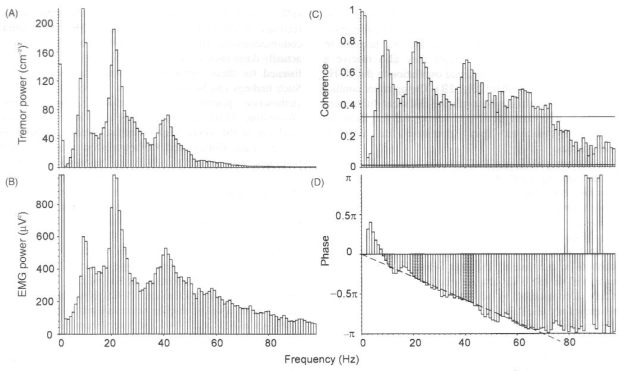
Power speectral estimates and coherence analysis of 50% maximum voluntary contraction of first dorsal interosseous muscle against an elastic resistance. There are peaks at 10, 22 and 41 Hz in accelerometer tremor record and rectified surface EMG power spectra. Coherence analysis reveals strong coherence especially at these peaks. Upper horizontal line is the 95% confidence interval for significantly greater coherence compared to the whole spectrum – only lower 100 Hz of spectrum shown. Lower horizontal line is 95% confidence interval for non-zero coherence. There is a constant linear phase lag of tremor behind EMG at all frequencies, indicating a value of 6.5 ms lag.
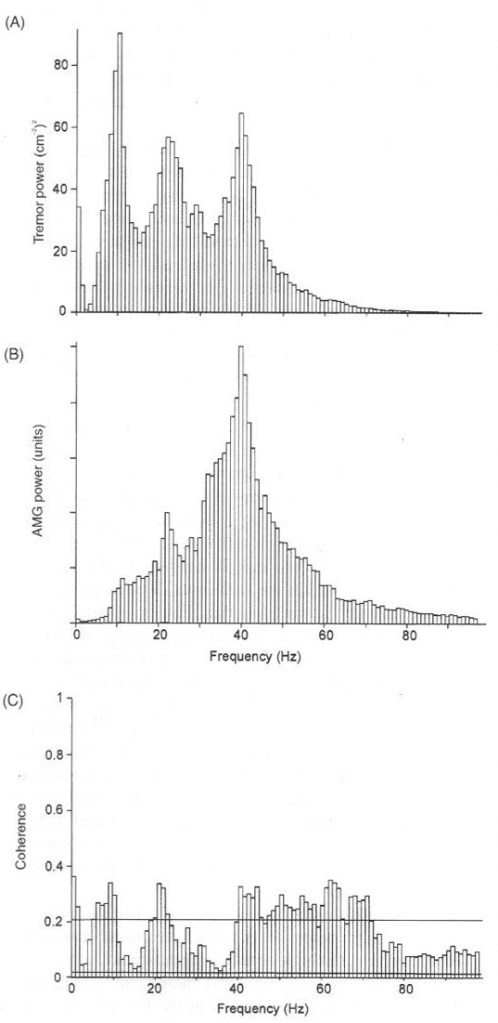
Same subject as in above figure under identical conditions. The acoustomyogram (AMG) is recorded by an electronic heart sounds monitor placed over the belly of first dorsal interosseous during contraction against elastic resistance. The “sounds” are generated directly by muscle activity as seen on the EMG power spectrum in previous slide. The microphone is not as sensitive at 10 Hz as 40 Hz, hence the larger 40 Hz peak compared with tremor and EMG.
It is temping therefore to speculate that the tinnitus of cervical spondylosis relates to overactivity of the sternomastoid muscles that originate just behind the external ear. Rather than the tinnitus sounds being imaginary or related to cochlear damage, patients are actually hearing their own muscles contracting. Certainly this interesting notion bears further investigation.
 Background
Background



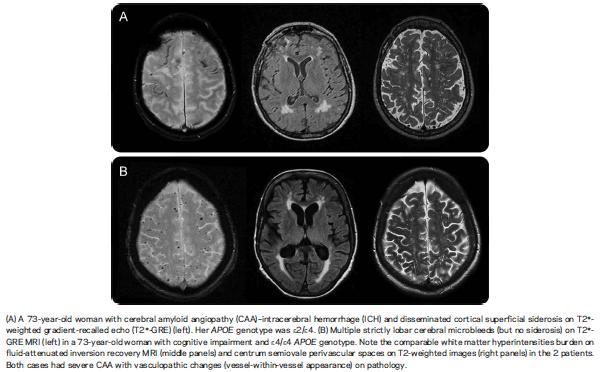

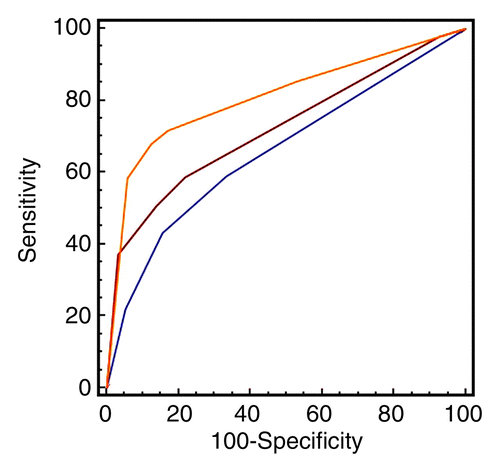
 Stroke, defined as a sudden vascular event resulting in localised brain damage (World Health Organisation, 1978), is without doubt a major challenge in health care, being the third most common cause of mortality in developed countries and the single greatest cause of lasting disability (Mant et al., 2004). In the UK, stroke patients occupy 2.6 million days in hospital beds a year, equivalent to one in five total acute hospital beds and one in four long-term beds (National Audit Office, 2005). Over the last decade, there have been increasing efforts to organise acute stroke care into dedicated stroke units and to raise public awareness that stroke is a medical emergency to be managed in a timely fashion (e.g. the FAST campaign).
Stroke, defined as a sudden vascular event resulting in localised brain damage (World Health Organisation, 1978), is without doubt a major challenge in health care, being the third most common cause of mortality in developed countries and the single greatest cause of lasting disability (Mant et al., 2004). In the UK, stroke patients occupy 2.6 million days in hospital beds a year, equivalent to one in five total acute hospital beds and one in four long-term beds (National Audit Office, 2005). Over the last decade, there have been increasing efforts to organise acute stroke care into dedicated stroke units and to raise public awareness that stroke is a medical emergency to be managed in a timely fashion (e.g. the FAST campaign).