 Migraine is one of the most common neurological conditions, and chronic migraine is a condition that, while less common than episodic migraine, is nevertheless a major cause of loss of quality of life in otherwise well individuals.
Migraine is one of the most common neurological conditions, and chronic migraine is a condition that, while less common than episodic migraine, is nevertheless a major cause of loss of quality of life in otherwise well individuals.
Once analgesia headache has been effectively treated, and tension type headache excluded, chronic migraine is treated with migraine preventative medications, often very effectively. However there are a proportion of patients who remain resistant to single or combination preventative treatments.
A novel target for migraine treatment is the calcitonin gene related peptide CGRP receptor on the smooth muscle of blood vessels in the head. CGRP is released from trigeminal ganglion efferents to the blood vessels to cause potent vasodilation as part of the trigeminovascular response (analogous to the “triple response” of pain, redness and swelling of skin inflammation). Blocking this may therefore block this response. Monoclonal antibodies raised against the receptor, or against CGRP itself, have been explored as migraine treatments.
This study describes a double blind trial on galcanesumab, one such monoclonal antibody targeting CGRP. The paper does not discuss the relative hypothetical or actual benefits versus other monoclonal Ab migraine therapies already marketed or in development.
Study Design
Around 270 patients were given each of two doses of galcanezumab by monthly subcutaneous injection, and 560 were given normal saline placebo. To be enrolled on the study, patients had to have 15+ headache days per month, at least 8 of which had to be migraine days. They needed at least 1 headache free day per month. If a patient failed >3 other preventatives, they were excluded. Before the study, patients had to stop all their existing migraine preventatives except propranolol or topiramate at least 30 days before study start.
Migraine days were defined as >30 minutes of migraine or probable migraine according to ICHD-3 beta criteria (even though the duration criterion of the latter is 4+ hours). If a patient thought it was a migraine and it did not satisfy the criteria but responded to a triptan, that also counted as a migraine day.
Over 90% of patients completed the study. Only 15% of patients were on topiramate or propranolol (not specified if this was the same proportion in the three treatment groups).
The primary outcome measure was migraine days per month. At the start of treatment, this was around 19 days. Placebo reduced this by 2.7 days per month, low dose galcanezumab by 4.8 days and high dose by 4.6 days. Therefore, compared to placebo, the drug on average reduced migraine by 2 days per month. There were only about 2 extra non migraine headache days per month on average.
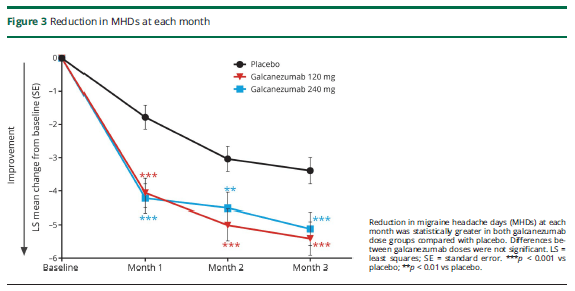
There were many secondary measures. Of note, 4.5% of placebo patients had a 75% reduction in migraine days, and 7% of low dose and 8.8% of high dose patients, while 0.5% of placebo patients had a 100% response, and 0.7% of low dose and 1.3 % of high dose patients (not significantly different).
There was no overall quality of life measure, but there was a migraine related quality of life measure that showed significantly more improvement, about 25% more improvement than placebo. There was a patient global disease severity 7 point scale, where there was a 0.6 point improvement from placebo, and 0.8 for low dose and 0.9 for high dose, only the latter reaching significance.
The side effect profiles were similar between placebo and drug, notably common in both groups! However, there were no concerning side effects, nor indeed any characteristic enough to tend to unblind the patients or investigators.
Opinion
The Journal Club thought it was strange that the study would exclude the very patients in whom the drug would mainly be used, namely those who had failed >3 conventional treatments. The focus was clearly on maximising benefit as measured by the study. By the same token, patients had to stop any preventatives before the study, even if they were partially beneficial, apart from topiramate and propranolol.
It was furthermore strange that only 15% of the recruited patients were on the two most common treatments for chronic migraine. Had they only been tried on the others, or had they had side effects? In real practice, there are usually at least some marginal benefits from preventatives and patients often remain on them.
It is therefore possible that many patients were treatment naïve as far as preventatives were concerned. This makes the 2 fewer migraine days per month vs placebo (from an initial 19 days per month) an all the more modest magnitude of benefit.
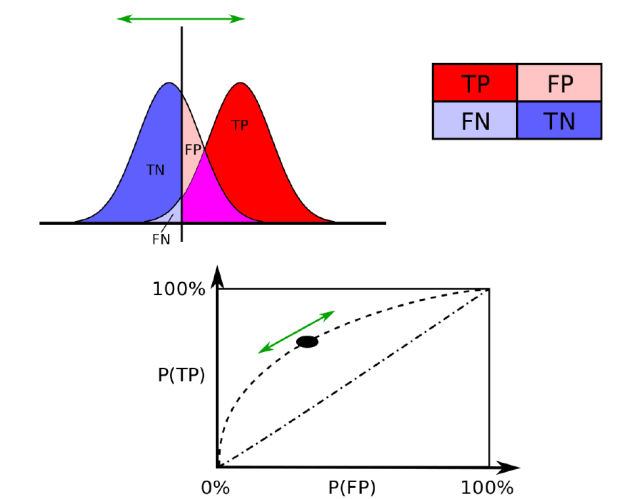
It is difficult to reconcile the cost of the drug with the fact that patients on average will still have 15 migraine days a month. Most patients would not consider this a treatment success, and certainly not such that a patient would happily be discharged from specialist care. In terms of patients having a 75%+ reduction in migraine days, generally the minimum level of meaningful benefit in a pain study, the excess over placebo was only 3-4% of patients.
The lack of a general quality of life measure means that cost benefit analysis cannot be performed. The quality of life measure used was specific for migraine and likely to show much larger differences; a cured migraine sufferer might have a near 0% to 100% swing on this scale, but another individual considering the range from death to total disability to perfect health might assign curing migraine only a swing from 90% to 100%.
A major aspect of migraine care is what happens when treatment is stopped. Patients do not want lifelong medication, let alone lifelong monthly injections. Fortunately we find that after six months of treatment, traditional preventatives can often be withdrawn. Although the study mentioned that there was an open label period and then a wash out period, we do not know any of these results; presumably they are to be held back for another publication. Is there rebound migraine on treatment withdrawal? Any funding body would want to know if the patients would likely need the treatment for 3-6 months or for many years.
As a final point, it was queried whether the definition of migraine is sufficiently specific; perhaps this limits the observed benefit in this and similar studies. Some headaches recorded as migraine may be tension type headache and therefore not responsive to specific anti-migraine treatment. The table below shows the relevant criteria.
ICHD-3 Headache Diagnostic Criteria
| Probable Migraine | Probable Tension Type Headache | Definite Tension Type headache |
| 2+ of: | 2+ of: | All of: |
| 4-72 hours duration | 30 min to 7 days duration | 30 min to 7 days duration |
| 2+ of:
Unilateral, Pulsing, Moderate+ severity, Avoid routine physical activity |
2+ of:
Bilateral Pressing or tightening Moderate- severity Not aggravated by routine activity |
2+ of:
Bilateral Pressing or tightening Moderate- severity Not aggravated by routine activity |
| Nausea or
Photo plus phonophobia |
No nausea
Not both phono and photophobia |
No nausea
Not both phono and photophobia |
A headache is diagnosed as a migraine if fits probable migraine and is not a better fit with another headache diagnosis, which presumably means definite rather than probable tension type headache. The severities and durations overlap so they cannot distinguish. One of photophobia or phonophobia overlaps. So a unilateral, pressing headache with avoidance of routine activity with no nausea no photophobia and no phonophobia is classified as migraine as long as it lasts 4 hours, but it seemed that some of the migraine days were half an hour of headache. Also a headache not satisfying these criteria is a migraine if there is a response to triptans, but we have seen the large placebo response already from the main data. In general practice a tension type headache might be unilateral, and might interfere with routine activity if at the more severe end of the scale; certainly a neck ache or jaw (including temporalis muscle) ache from which a tension headache may arise may have these features.
The paper on which this Journal Club article is based was presented by Dr Piriyankan Ananthavarathan, Specialist Registrar in Neurology at Barking, Havering and Redbridge University Hospitals Trust.



 Stroke is the most common cause of disability in Western Countries, and its lifetime risk is 1 in 6 for men and 1 in 5 for women. While managing acute stroke patients in hyperacute stroke units overall has modest benefits for short and long term outcome (e.g. 51% versus 47% independence and 29% versus 33% mortality), specific therapeutic options are limited. The first major option for treatment of ischaemic stroke was intravenous thrombolysis, paralleling its previous development in acute myocardial infarction.
Stroke is the most common cause of disability in Western Countries, and its lifetime risk is 1 in 6 for men and 1 in 5 for women. While managing acute stroke patients in hyperacute stroke units overall has modest benefits for short and long term outcome (e.g. 51% versus 47% independence and 29% versus 33% mortality), specific therapeutic options are limited. The first major option for treatment of ischaemic stroke was intravenous thrombolysis, paralleling its previous development in acute myocardial infarction. Introduction
Introduction Background
Background Background
Background
