Summary for General Readers
Summary for General Readers
As discussed in the accompanying primer, I chose to review a research article (Weber-Carstens et al., 2010) I found that looked both at risk factors for development of critical illness myopathy and a new diagnostic test for it.
The premise of the test is this; traditionally both nerve and muscle diseases are investigated electrophysiologically by inserting a tiny needle into a muscle and recording the electrical potential that occurs across the muscle when the nerve to the muscle is stimulated by a small electrical current applied through the skin over the nerve (this is only a little uncomfortable even for a wide awake patient). If there is a shrinkage in the recorded potential due to damage, there are other clues that indicate whether it is likely to be the nerve or the muscle that is the problem. But in an unconscious patient who may have two overlapping pathologies as described above, we need any extra information we can get. The new test actually stimulates the muscle directly, not the nerve, without needing voluntary co-operation on the part of the patent and records the muscle membrane excitability. Thus, this will be abnormal in a myopathy (e.g. a critical illness myopathy) but normal in a neuropathy (e.g. if the patient was in an intensive treatment unit (ITU) for Guillain Barre syndrome or coincidentally had diabetic neuropathy).
The study followed 40 patients who had been admitted to ITU and who had been broadly selected as being at high risk because they had persistently poor scores on basic life-functions (e.g. conscious level, blood pressure, blood oxygenation levels, fever, urine output). They looked at all the parameters that could put patients at risk of developing critical illness myopathy and then analysed these against the muscle membrane excitability test measurements. It was found that 22 of the patients showed abnormalities on this test, and these patients did indeed have more weakness and require a longer ITU stay, suggesting they had critical illness myopathy. In terms of factors that would predict development of myopathy, there was an important correlation between abnormal muscle membrane test findings and a certain blood test (raised interleukin 6 level) that indicates systemic inflammation or infection. Other (possibly overlapping) correlations included the overall disease severity, the overt presence of infection, a marker indicating resistance to the hormone insulin (IGFBP1), the requirement for adrenaline (called epinephrine in the US) type stimulants and the requirement for heavier sedation.
The study’s strengths are that it highlights an important area of patient management that may often be somewhat neglected, it seems thoroughly conducted with a convincing result and it not only describes a new test but shows how it may be clinically useful and validates it against the patients’ actual clinical outcome. I felt that a possible missed opportunity was relying solely upon the notoriously insensitive Medical Research Council (MRC) strength assessment system. At the levels they were recording (from around 2 to 4), the test is a bit better, however, and it at least reflects something that is clinically relevant. Values for the actual numbers of patients who were clinically weak such as to delay recovery in the test-positive vs test-negative patients would have been helpful. A quantitative limb strength measure (when the patient later wakes up more fully) or a measure of respiratory efforts might also have been useful. Finally, one cannot take the proportion of patients with critical illness myopathy on this test as a prevalence level (though the authors do not purport to do this). This is because a positive test result does not necessarily indicate a clinically significant myopathy, as mentioned above, and because the patients were already selected as being severe cases. A study looking at any ITU patients would be interesting; for example would there be certain risk factors for myopathy even in patients who were otherwise generally less critically ill?
This question brings me to another point that I think may be important. After reviewing the journal, further review of the wider literature on critical illness myopathy led to my understanding that there are three distinct pathological types (meaning appearances under microscopy and staining), but to a variable extent they may all be caused by the catabolic state of the ill patient. A catabolic state means a condition where body tissues are broken down for their constituent parts to supply glucose for energy or amino acids to make new protein. In a critically ill patient, the physiological response is to go “all out” to preserve nutrition for vital organs, such as the brain, the heart and the internal organs, in the expectation that there will be little or no food intake. Especially if the patient has fever or is under physiological stress, there is also an increased demand for nutrition. So the body breaks down the protein of its own tissues for its energy supply, and the most plentiful source for this “meat”, as with any meat we might eat, is… muscle. My accompanying journal club review goes beyond the research article to look at measures to limit or correct this “self-cannabilistic” tendency in ITU patients.
But related to the issue described above regarding selection of patients are some intriguing questions. What if the same phenomenon occurred to a lesser extent in other patients who were sick but not severely enough to need transfer to ITU? What would be the effect if a patient were in a chronic catabolic state already because they were half-starved as the consequence of a neurological problem that affected the ability to swallow, or if they already had a muscle-wasting neurological condition?
It is possible, for example, that this could have a major impact on care of patients suffering from acute and not so acute stroke. Identifying and specifically treating those whose weakness is not only due to their stroke but to a superadded critical illness myopathy induced by the fact that they are generally very unwell, susceptible to infection and poorly nourished due to swallowing problems could have a significant positive influence on rate of recovery and final outcome.
Scientific Background
Introduction
Critical illness myopathy is a relatively common complication experienced by patients managed in intensive care, occurring in 25-50% of cases where there is sepsis, multi-organ failure or a stay longer than seven days. I chose a research article on this condition for online journal club review because I had previously assumed the condition was rare and knew little about it until the fact that a patient of mine was identified as having the condition prompted me to engage in some background reading. The study I have reviewed focuses on diagnosis and prediction of risk factors. As a Neurologist I was particularly concerned with difficulty in diagnosis when the reason for the patient requiring ITU management in the first place is that they have a primary neuromuscular disorder. In other words, the critical illness myopathy is a superadded cause for their weakness. First, I describe some of the general background on this seldom-reviewed (by me at any rate!) condition.
Epidemiology
The exact incidence of critical illness myopathy even in the well-defined situation of ITU, is unclear and varies between studies, perhaps reflecting different case mixes and difficulty distinguishing from critical illness polyneuropathy. Indeed in some cases, myopathy and neuropathy may coexist. An early prospective study by Lacomis et al. (1998) found electromyographic (EMG) evidence of myopathic changes in 46% of prolonged stay ICU patients. When looking at clinically apparent neuromyopathy, De Jonghe et al. (2002), found an incidence of 25%, with 10% of the total having EMG and muscle biopsy evidence of myopathic or neurogenic changes. In a review by Stevens et al. (2007), the overall incidence of critical illness myopathy or neuropathy was 46% in patients with a prolonged stay, multi-organ failure or sepsis. A multi-centre study of 92 unselected patients found that 30% had electrophysiological evidence for neuromyopathy (Guarneri et al., 2008). Pure myopathy was more common that neuropathic or mixed types and carried a better prognosis, with three of six recovering fairly acutely and a further two within six months.
Investigation
In a patient with limb weakness in an intensive care setting there should be a high level of suspicion for critical illness neuromyopathy. Nerve conduction studies (NCS) and EMG may help to distinguish critical illness polyneuropathy with more distal involvement and large polyphasic motor units on EMG, from critical illness myopathy with more global involvement, normal sensory nerve conduction and small polyphasic units.
However, there remain potential difficulties. First, EMG is easier to interpret when an interference pattern from voluntary contraction can be obtained, but this might prove impossible with a heavily sedated or comatose patient. Second, when the patient’s primary condition is neurological, such as in Guillain Barre syndrome, myasthenia, myopathy or motor neurone disease, it may be difficult to distinguish NCS and EMG abnormalities of these conditions from those of superadded critical illness.
In cases of suspected critical illness myopathy, the most definitive investigation is muscle biopsy. Histologically, it manifests in one of three ways, and these may be distinguished from neurogenic changes or other myopathic disease.
Subtypes of Critical Illness Myopathy: Minimal Change Myopathy
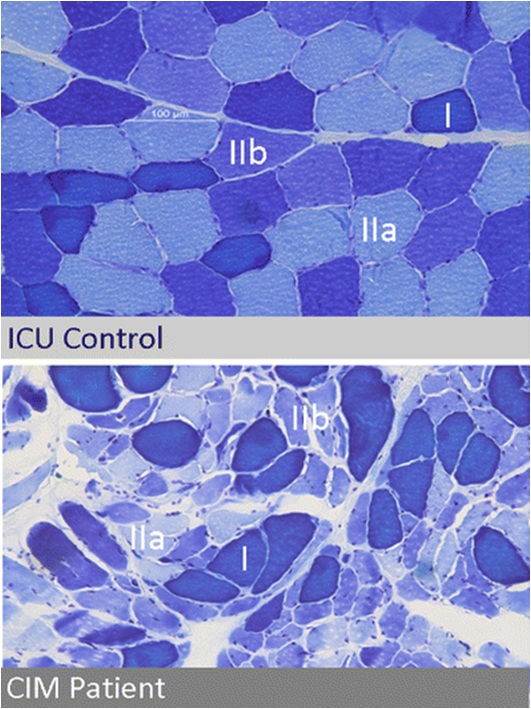
 The first subtype is minimal change myopathy. There is increased fibre size variation, some appearing atrophic and angulated as they become distorted by their normal neighbours. Type II fibre involvement may predominate, perhaps because fast twitch fibres are more susceptible to fatigue and disuse atrophy. There is no inflammatory response and thus serum creatine kinase is normal.
The first subtype is minimal change myopathy. There is increased fibre size variation, some appearing atrophic and angulated as they become distorted by their normal neighbours. Type II fibre involvement may predominate, perhaps because fast twitch fibres are more susceptible to fatigue and disuse atrophy. There is no inflammatory response and thus serum creatine kinase is normal.
Clinically, it may be apparent only as an unexpected difficulty weaning from ventilation, and the EMG changes may be mild, making muscle biopsy more critical.
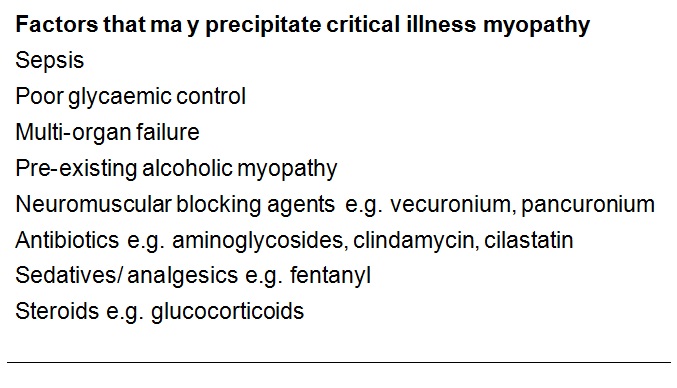
The condition may lie on a continuum with disuse atrophy, but made more extreme by a severe catabolic reaction induced by sepsis and systemic inflammatory responses triggering multi-organ failure (Schweickert & Hall, 2007). Muscle is one such target organ; ischaemia and electrolyte and osmotic disturbance in the critically ill patient trigger catabolism by releasing glucocorticoids and cytokines such as interleukins and tumour necrosis factor. For example, Interleukin 6 promotes a high affinity binding protein for insulin like growth factor (IGF) to down-regulate the latter and thereby block its role in glucose uptake and protein synthesis. This is paralleled by a state of insulin resistance. Muscle may be particularly susceptible to catabolic breakdown, being a ready “reserve” for amino acids to be used in proteolysis to maintain gluconeogenesis for other vital tissues in the body’s stressed state (Van den Berghe, 2000). A starved patient may lose around 75 g/day of protein, while a critically ill patient may lose up to 250 g/day, equivalent to nearly 1 kg of muscle mass (Burnham et al., 2003). Disuse, exacerbated iatrogenically by sedatives, membrane stabilisers and neuromuscular blocking drugs, may impair the transmission of myotrophic factors and further potentiate the tendency to muscle atrophy (Ferrando, 2000).
Subtypes of Critical Illness Myopathy: Thick Filament Myopathy
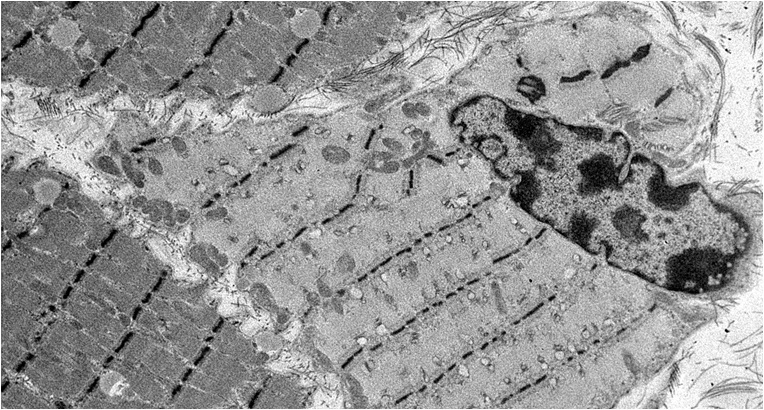
 The second histological subtype is thick filament myopathy. There is selective proteolysis of myosin filaments, as seen by smudging of fibres on Gomorri Trichrome light microscopy and directly on electron microscopy. Since myosin carries the ATPase moiety, this is apparent on light microscopy as a specific lack of ATPase staining of both type I and type II fibres. Clinically, patients may have global flaccid paralysis, sometimes including ophthalmoplegia, and difficulty weaning from the ventilator. The CK may be normal or raised. Thick filament myopathy appears to have a similar pathophysiology to minimal change myopathy, but may be especially associated with high-dose steroid administration and neuromuscular blocking agents, particularly vecuronium.
The second histological subtype is thick filament myopathy. There is selective proteolysis of myosin filaments, as seen by smudging of fibres on Gomorri Trichrome light microscopy and directly on electron microscopy. Since myosin carries the ATPase moiety, this is apparent on light microscopy as a specific lack of ATPase staining of both type I and type II fibres. Clinically, patients may have global flaccid paralysis, sometimes including ophthalmoplegia, and difficulty weaning from the ventilator. The CK may be normal or raised. Thick filament myopathy appears to have a similar pathophysiology to minimal change myopathy, but may be especially associated with high-dose steroid administration and neuromuscular blocking agents, particularly vecuronium.
Subtypes of Critical Illness Myopathy: Acute Necrotising Myopathy
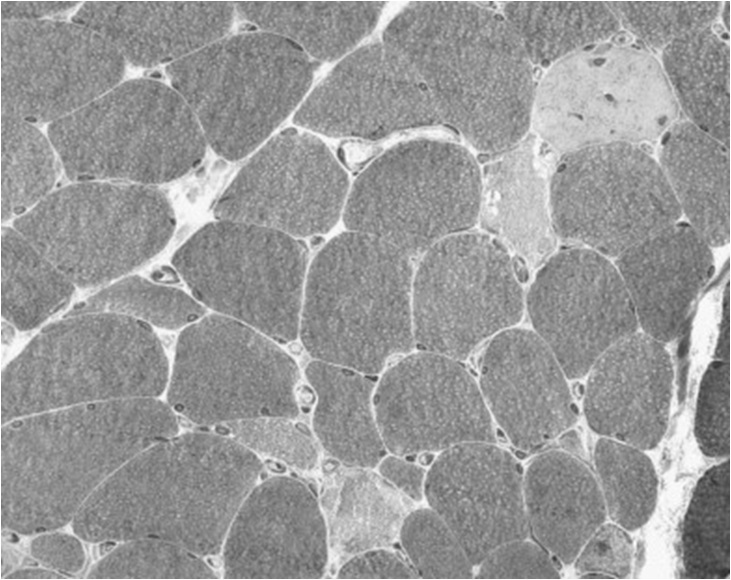
 This is a more aggressive myopathy, with prominent myonecrosis, vacuolization and phagocytosis. Weakness is widespread and the CK is generally raised. Its aetiology may relate to the catabolic state rendering the muscle susceptible to variety of additional, possibly iatrogenic, toxic factors. It may lie on a continuum with, and progress to, frank rhabdomyolysis.
This is a more aggressive myopathy, with prominent myonecrosis, vacuolization and phagocytosis. Weakness is widespread and the CK is generally raised. Its aetiology may relate to the catabolic state rendering the muscle susceptible to variety of additional, possibly iatrogenic, toxic factors. It may lie on a continuum with, and progress to, frank rhabdomyolysis.
Management
There are a number of steps in managing critical illness myopathy.

- First, iatrogenic risk factors should be identified and avoided where possible (see list above).
- Second, appropriate nutritional supplementation may be helpful but objective evidence for this is sparse. Parenteral high dose glutamine supplementation may improve overall outcome and length of hospital stay (Novak et al., 2002), and since critical illness myopathy is so common at least some of this may be by partly reversing the catabolic tendency in muscle. Other amino acid supplements and antioxidant supplements (e.g. glutathione) could have similar effects but have not been adequately trialed. There is again no conclusive proof in favour of androgen or growth hormone supplements, and in the latter case there may be adverse effects (Takala J et al., 1999). Tight glucose control with intensive insulin therapy reduces time on ventilatory support, and may protect against critical illness neuropathy, but the effect on myopathy is not clear (van den Berghe et al., 2001).
- Finally, early physiotherapy encouraging activity may be helpful, as shown in a randomised controlled trial (Schweickert et al., 2009), perhaps preventing the amplification of catabolic effects by lack of activity.
Journal Review
The research article reviewed here (Weber-Carstens et al., 2010) describes a study looking at a relatively new electrophyiological test for myopathy, namely measurement of muscle membrane electrical excitability to direct muscular stimulation. An attenuated response on this test will indicate a myopathic process unlike a reduced traditional compound muscle action potential that could reflect either neural or muscular pathology. Furthermore, while an EMG interference pattern is dependent on some background ongoing voluntary muscle activity, the test can be performed on a fully unconscious patient. The study uses this test to explore the value of various putative clinical or biochemical markers recorded early in the patient’s time on ITU that might subsequently predict the development of critical illness myopathy.
There were 40 patients selected for study on the basis that they had high (poor) Simplified Acute Physiology (SAPS-II) scores for at least three days in their first week on ITU. It was found that 22 of these subsequently had an abnormally muscle membrane excitability. As was also shown in a previous study, the abnormal test values in these patients corresponded to a clinical critical illness myopathy state in that they were weaker than the others on clinical MRC strength testing and they also took significantly longer to recover as measured by ITU length of stay.
The main finding was that multivariate Cox regression analysis pointed to blood interleukin 6 levels as an independent predictor of development of critical illness myopathy, as was the total dose of sedative received. However the predictive value of this correlation on its own was modest. In an overall predictive test combining a cut-off level of Il-6 of 230 pg/ml or more and a Sequential Organ Failure Assessment (SOFA) score of 10 or more at day 4 on ITU, the observed sensitivity was 85.7% and specificity 86.7%. There were also other potentially co-dependent predictive risk factors, including markers of inflammation, disease severity, catecholamine use and IGF binding protein level. Higher dose steroids, aminoglycosides and neuromuscular blocking agents were interestingly not associated with critical illness myopathy in this sample.
Opinion
The study is clearly described and carefully conducted. The electrophysiological test appears to have real value, and is perhaps something that should be more widely introduced as a screening test before a muscle biopsy, given the latter test’s potential complications. The test can also be performed at a relatively early stage on a completely unconscious patient, where interventions to address the problem can be made in a more timely manner. Certainly I am going to discuss the feasibility of this test with my neurophysiological colleagues.
As the authors point out, perhaps the fact that they only recorded blood tests such as Interleukin levels on two occasions per patient meant that they missed the true peak level in some patients – its predictive value might otherwise have been stronger. I would have liked to have seen a more explicit link between their muscle membrane excitability and clinically relevant weakness. They show a reduction in mean MRC strength grade from around 4 to 2, which is clinically meaningful at these strength levels, but objective strength testing or respiratory effort measurements would have been advantageous, as well as the actual numbers of patients who were clinically severely weakened rather than just those with abnormal electrophysiology.
I think further study on unselected patients is important, even if it means that perhaps only 22 out of 100 rather than 22 out of 40 will have abnormal electrophysiology. This is because it might not only be those patients selected for the study on the basis of persistently poor physiology scores who could develop critical illness myopathy. A predictive marker in otherwise low risk patients might prove even more useful.
By way of general observation rather than opinion on this research, and extending the argument on investigating less critically ill patients, I have wondered if critical illness myopathy might in fact occur in acutely unwell patients who do not reach ITU at all. There are many neurological and other conditions that predispose to catabolic states, such as patients with chronic infection or inflammation, those who had preexisting disuse atrophy, those on steroids, or those who were already chronically malnourished due to poor care or poor or unsafe swallowing before they deteriorated such that they required acute hospital care. Even patients without pre-existing disease, such as those who have suffered acute stroke, may subsequently be susceptible to a catabolic state due to aspiration, other infection, immobility or suboptimal nutrition. One can speculate that large numbers of patients with stroke, multiple sclerosis relapse or other acute deteriorations requiring neurorehabilitiation may have significantly impaired or delayed recovery due to unrecognized superadded critical illness neuropathy. Certainly in stroke, important measures found to improve outcome, such as early physiotherapy and mobilisation, early addressing of nutrition, treating infection and good glycaemic control, happen to be among the key elements in treating critical illness myopathy. More directed and aggressive management along these lines in a subgroup of these patients who have markers for critical illness myopathy might further accelerate improvement and achieve a better final outcome.
 Review for General Readers
Review for General Readers